
Microbial Solutions
|
Digging Deeper into Sequences and Strains of Microorganisms, Part II
How inferred multilocus sequence typing using whole genome sequencing can obtain strain-level resolution
Species identification (ID) and strain typing are both key components of a comprehensive environmental monitoring program and necessary for investigations into contamination events. When these events occur, it is critical to identify the source of contamination so effective corrective and preventive actions can be implemented.
Both MALDI-TOF and Sanger-based rRNA gene sequencing can identify organisms down to the species level. However,a species-level ID alone is not going to narrow down the possibilities of where or how many sources the isolates are coming from. This task requires a higher resolution method to differentiate the isolates at the strain level allowing for comparability to other samples collected during the investigation. Strain typing generates more appropriate data to look at relationships within a species and can help focus the investigation to specific areas within a site to find the source of contamination and determine the root cause.
Comparing genetic data is key to IDing the contamination source
Strain-level resolution using inferred multilocus sequence typing (MLST) is conducted by analyzing and comparing sequence variations in protein coding genes for each strain of a particular bacterial or fungal species under investigation. These genes contain more sequence variability compared to the rRNA gene regions used for ID providing a greater degree of discriminatory power necessary for strain typing applications. There are many internationally recognized public databases that define strain typing schemes for well over 150 microorganisms. These schemes list the gene regions of interest and follow internationally standardized content and nomenclature. The sequence information from all the genes or gene fragments is directly compared to understand the relatedness between the isolates. Investigations can be closed more quickly and efficiently by elucidating strain-level similarities or differences between isolates thus providing precise information on contamination sources.
Both Sanger sequencing and next-generation sequencing (NGS) can be used for MLST. Next generation sequencing methods take a very different technological approach to generating data and accomplish this in a massively parallel process. The NGS-based MLST approach uses whole genome sequencing (WGS) where, quite literally, the sequence for the entire genome of the organism of interest is determined. Different data analysis pipelines are used for the different sample types and applications. In this case, after generating the genome sequence for each isolate, the sequences of the MLST gene targets, as determined by the public databases, are inferred by bioinformatically analyzing the entire genome, eliminating the use of multiple primer sets, and individual PCR/sequencing reactions. The workflow for MLST via NGS is shown in Figure 1.
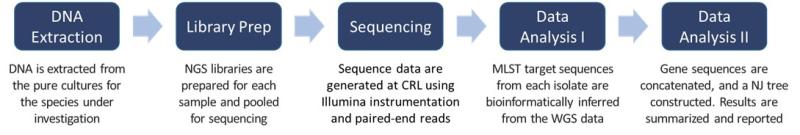
In the example below, after isolating genomic DNA from 21 fungal isolates, NGS was used to generate the WGS. The sequences for each of six published gene targets were inferred from the WGS data for each of the isolates that were part of the investigation.
Strain relatedness was visualized by constructing a Neighbor-Joining (NJ) phylogenetic tree based on the concatenated sequences for all protein coding gene targets (Figure 2). An analysis of the branching pattern of the NJ tree illustrated three distinct groups of strain clusters for these fungal samples, suggesting the existence of three different sources of the isolate contributing to the contamination event.

With these data, the company could identify sources of contamination, track areas common to each strain, track how that organism spreads through the environment, and eliminate the sources of contamination.
Although the protein coding genes, and by extension the bioinformatic data analysis pipeline, used for MLST analysis will vary by species, the steps involved in generation of whole genome sequence data remains the same for each organism. Simplifying the process. This approach can provide strain typing for any microorganism as long as protein coding genes for the species are well-defined in published literature.
The Sanger method is perfect for sequencing parts of genes, one fragment at a time. By sequencing millions of fragments in parallel, NGS can easily generate data for entire genomes. You can even bypass specific MLST schemes and compare genome sequences to each other looking for overall relatedness or even small nucleotide differences. Whatever the method used for generating sequence data – these high-level comparison studies at the strain level are critical for investigations into when a manufacturing process has gone awry.