Discovery
|
David Clark, PhD
Hop To It!
How to jump from one molecule to another, fast!
For those who might not be familiar with it, the phrase “Hop to it!” means “act or move quickly”. Speed is often of the essence in drug discovery where competition to bring a drug to market is often fierce and the potential rewards for being first to market (or not too far behind) can be enormous.
So, in the early stages of drug discovery, it can be invaluable to have rapid methods for proposing novel compounds for synthesis and testing. One such method is known as “scaffold hopping” (sometimes also referred to as “lead hopping” or “core hopping”).
In medicinal chemistry and computer-aided drug design, we often spend most of our time working on what we call a “series” of compounds, that is usually defined by having a common scaffold or core. In this situation, we are interested in finding substituents to attach to the scaffold to improve the properties of the potential drug compound (Figure 1). For instance, we might be seeking to enhance the potency of the compound for its biochemical target or to improve its physicochemical or pharmacokinetic properties.
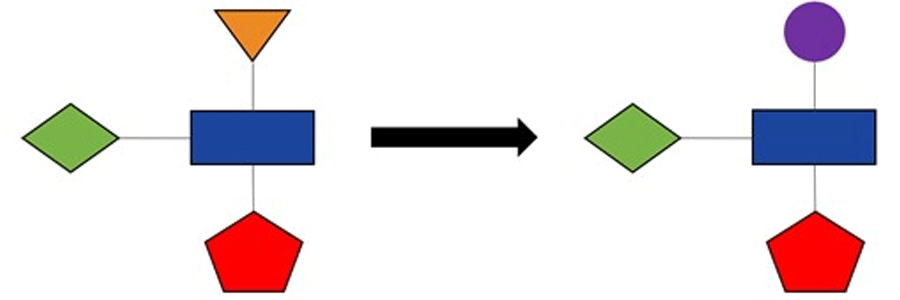
Figure 1: Optimising compound properties through modification of substituents. In this schematic illustration, the scaffold/core is represented by the blue rectangle and the substituents by other coloured shapes.
However, occasionally, we need to identify a novel scaffold – and often quickly—perhaps to obtain an intellectual property position, or to overcome some liability that has been discovered with the current scaffold. This brings us into the realm of scaffold hopping, the topic of this post (Figure 2). The aim of scaffold hopping is to replace the core of the compound with a different chemical structure while retaining the geometry of the attached substituents, some, or all of which might be making key contributions to the binding to the protein.
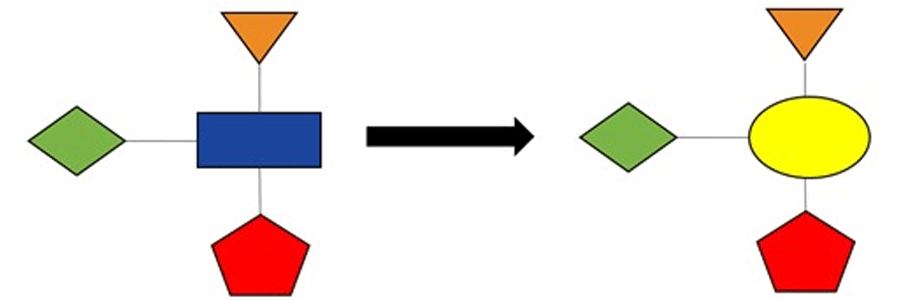
Figure 2: Schematic illustration of scaffold hopping, where the scaffold (blue rectangle) in the left-hand compound is swapped for a replacement (yellow ellipse) in the right-hand compound.
The process of scaffold hopping can, of course, be carried out without the application of computational methods, relying instead on the experience and expertise of medicinal chemists. However, algorithmic approaches, coupled with large structural databases, offer the possibility of rapidly assessing, and identifying, potential scaffold replacements that might not be otherwise considered.
Such is the importance and popularity of scaffold hopping that all the main vendors of computer-aided drug design (CADD) software market tools for it. These include BROOD (OpenEye), Core Hopping (Schrödinger), Spark (Cresset), SHOP (Molecular Discovery) and ReCore (BiosolveIT). An illustrative example of the use of the latter piece of software by scientists at Roche is shown in Figure 3.
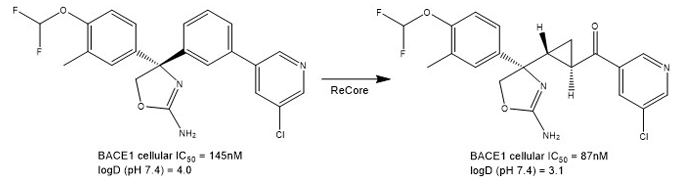
Figure 3: Real-world example of scaffold hopping taken from a project at Roche targeting the BACE-1 enzyme.
The Roche group was engaged on a project seeking to design inhibitors of the enzyme, BACE-1, which has been implicated in Alzheimer’s disease. The team was seeking to improve the solubility of its compound series by reducing lipophilicity (measured as logD at pH 7.4). The ReCore program suggested the replacement of the central phenyl ring in the left-hand side compound by a transcyclopropylketone moiety. When the resulting right-hand side compound was synthesised and tested, it was found to have a significantly reduced logD value and a concomitantly improved solubility while still maintaining excellent potency. When the two inhibitors were co-crystallised with BACE-1, the effectiveness of the scaffold hop was clearly illustrated (Figure 4).
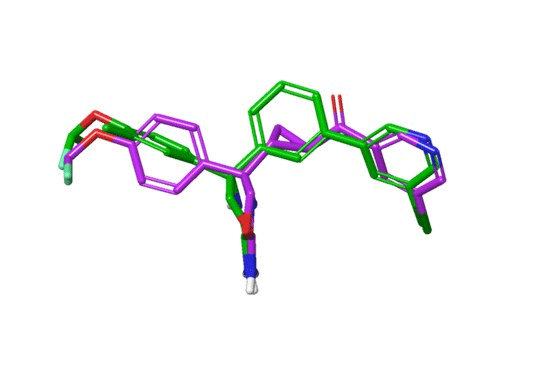
Figure 4: X-ray poses of BACE-1 ligands from RCSB database entries 5EZZ and 5EZX.
More recently, as part of a long-term collaboration between Charles River and Chiesi Farmaceutici, a novel core-hopping workflow, combining brute-force enumeration with shape screening, followed by other computational filters, was used to design a novel inhibitor of the kinase ROCK1. The workflow was applied to an X-ray structure of ROCK1 complexed with an inhibitor developed by Vertex Pharmaceuticals and led to the discovery of a novel compound containing a seven-membered azepinone ring (Figure 5).
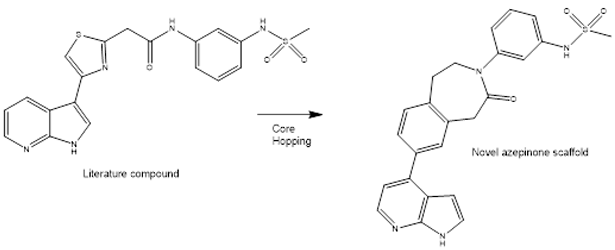
Figure 5: Real-world example of scaffold hopping taken from a collaboration between Charles River and Chiesi Farmaceutici.
Looking at the 2-D structures in Figure 5, the power of the scaffold hopping is not immediately apparent. However, if we look at the X-ray poses of the literature compound and an example of an azepinone compound bound to ROCK, the effectiveness of the scaffold hop is clear (Figure 6). The hinge-binding azaindole and P-loop binding phenyl moieties at either end of the compounds overlay very closely, even though the connecting pieces between them are completely different. In addition, the amide carbonyl of the literature compound and the carbonyl of the azepinone lie close in space and can form the same hydrogen bond with the protein.
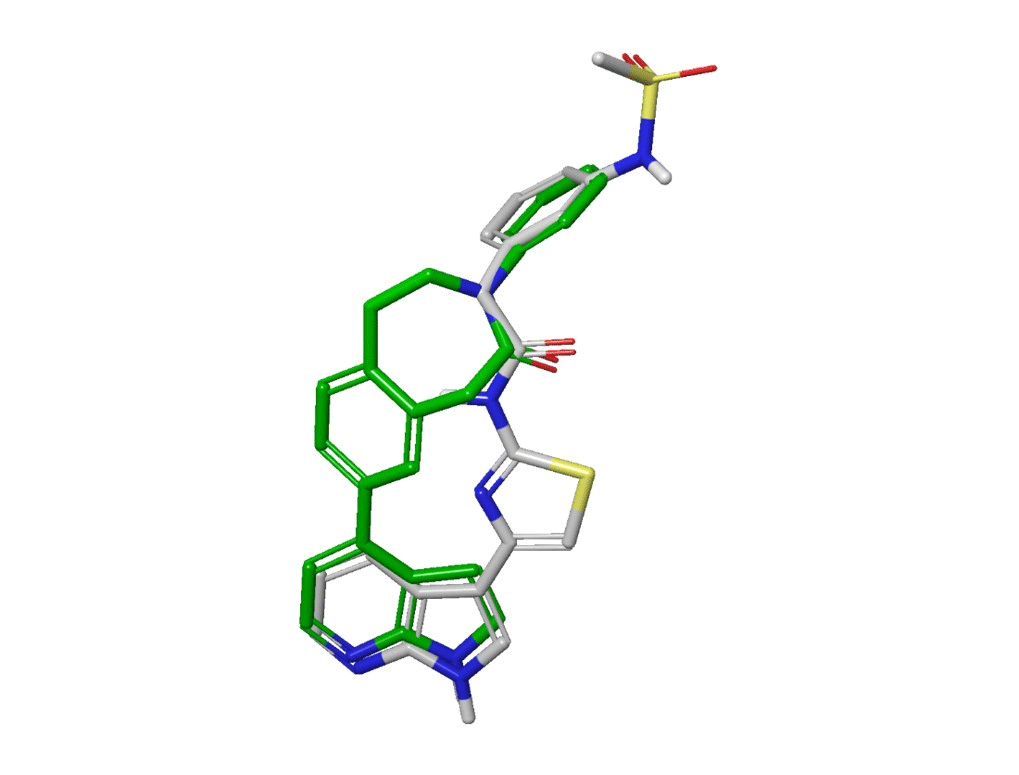
Figure 6: Ligand poses taken from RCSB X-ray structures 5KKS (white) and 9EP8 (green).
Of course, scaffold hopping is only one tool amongst many in the CADD toolbox that can be applied to accelerate drug discovery programs. For more information, please check out the CADD pages on the Charles River website.

Discovery Services
Now, more than ever, the research community is faced with an evolving landscape paved with new challenges. We know your work must continue for the patients who are waiting on new therapies. Our Discovery team is prepared and equipped to support you through these challenging times and keep your research moving forward.



