Discovery
|
Paul Wan, PhD
First-of-its-Kind Crystal Structure
Ancient endogenous retroviral protein reveals surprising structural clues that can lead to development of novel therapies
 It is surprising to learn that 54 percent of the human reference genome is comprised of repetitive elements. Some of these repeats are relics of past retroviral infections that occurred throughout human evolution. Most of these insertions are unable to generate active proteins. However, a subset of human endogenous retroviruses (HERVs) can express proteins with full reverse transcriptase (RT) activity. Expression of these HERV RTs has been associated with a variety of diseases, including cancer, although a direct link has yet to be established. This observation has inspired several clinical studies of antiviral RT inhibitors in these indications.
It is surprising to learn that 54 percent of the human reference genome is comprised of repetitive elements. Some of these repeats are relics of past retroviral infections that occurred throughout human evolution. Most of these insertions are unable to generate active proteins. However, a subset of human endogenous retroviruses (HERVs) can express proteins with full reverse transcriptase (RT) activity. Expression of these HERV RTs has been associated with a variety of diseases, including cancer, although a direct link has yet to be established. This observation has inspired several clinical studies of antiviral RT inhibitors in these indications.
The Charles River Structural Biology team in Early Discovery was part of an initiative led by ROME Therapeutics that determined the X-ray structure of an HERV-K (HML-2 [human endogenous MMTV-like] subtype) reverse transcriptase (see image above). Findings from this initiative appeared recently in the Proceedings of the National Academy of Sciences.
This structure has not been modeled previously and can inform the design of selective HERV-K RT tool molecules for drug target validation. The work has answered some key questions about the structural similarities between HERV-K RT and other viral RTs, as well as why some RT inhibitors effective against HIV-1 RT do not block HERV-K RT.
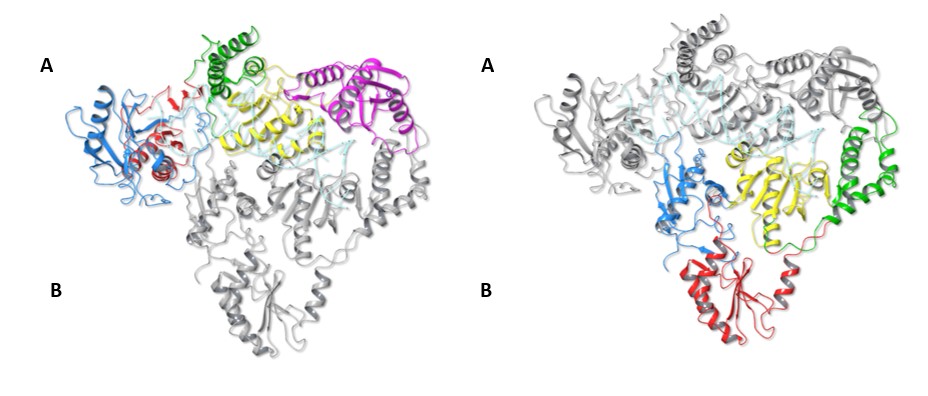
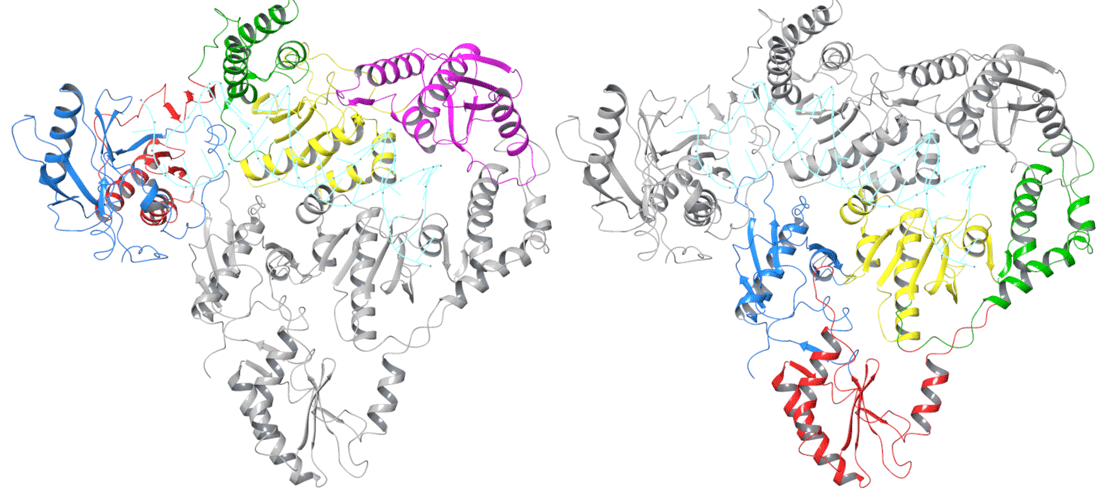
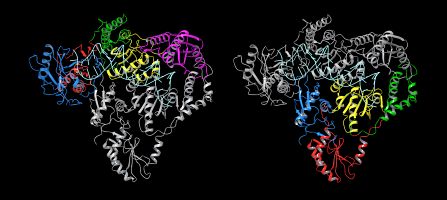
Fig. 1 The Structure of HERV-K RT Dimer
Figure legend: HERV-K RT adopts a dimer structure, comprised of molecule A and molecule B. Molecule A is coloured in the left image and molecule B the right. The subdomain organization of HERV-K RT follows that of HIV-1 RT. The polymerase domain is composed of three subdomains: fingers (blue), palm (red), and thumb (green). The polymerase domain is linked to the C-terminal RNase H domain (magenta) by the connection subdomain (yellow).
Despite sharing higher sequence identity with the monomeric MMTV RT (45% identity) vs HIV-1 RT (28% identity), HERV-K RT is an asymmetric homodimer with subdomain arrangement and interactions that are reminiscent of HIV-1 RT (figure 2). In fact, the images of our HERV-K RT structure overlaid with that of HIV-1 RT reveal striking structural similarities of the two proteins (figure 3).
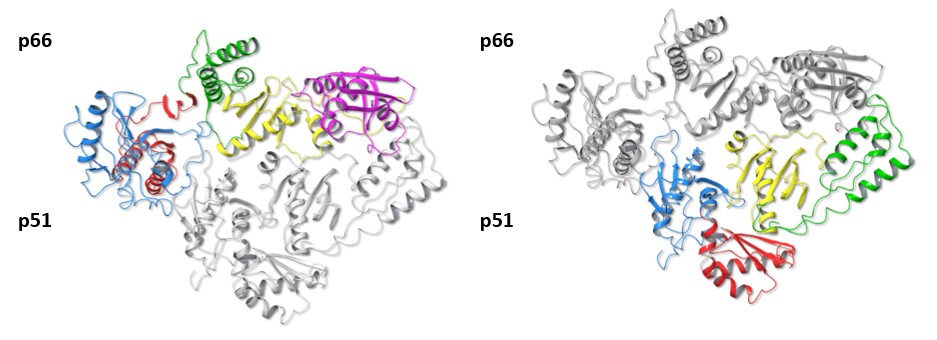
Fig. 2 The Structure of HIV-RT p66 and p51 Dimer
The structure of HIV-1 RT with the upper colored p66 subunit on the left and the p51 subunit coloured on the right. The subdomains are colored as for Figure 1.
The two molecules of the HERV-K RT dimer adopt arrangements similar to those observed in the heterodimeric HIV-1 RT structure (p66/p51). Molecule A of HERV-K RT adopts the active polymerase + RNase H configuration of the larger p66 subunit, which contains the full-length HIV-1 RT sequence. HERV-K RT’s molecule B in turn adopts an alternative folding similar to the shorter HIV-1 RT p51 subunit.
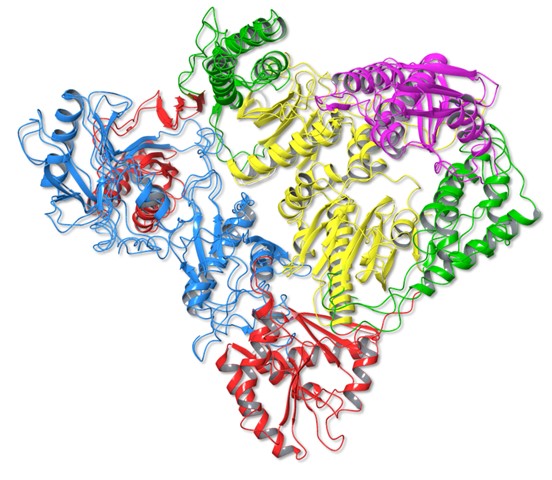
Fig. 3 Superimposition of HERV-K RT and HIV-1 RT
When HERV-K RT and HIV-1 RT are superimposed, the dimer structures and subdomain arrangements can be seen to be almost identical, colors depicting each domain as in Figures 1 and 2

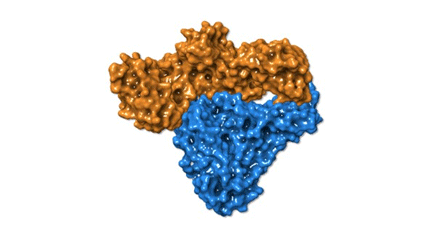
Fig. 4 Space-Filling Models of HERV-K RT and HIV-1 RT
Another perspective can be seen with space filling models of the same structures: HERV-K RT molecule A (orange) molecule B (blue), HIV-1 RT p66 subunit (green) p51 subunit (purple).
Why does inhibition of HERV-K RT and HIV-1 RT by certain RT inhibitors (RTIs) differ?
While the active site residues of HIV-1 RT and HERV-K RT are very similar, there are key differences that impact NRTI action. This is best illustrated with the HIV-1 RT crystal structure bound to the familiar AIDS drug AZT, which blocks both HIV-1 RT and HERV-K RT. The structure shows that protein residues surrounding the active component of AZT are identical between HIV-1 RT and HERV-K RT. The structural differences occur in the area termed the ‘Gate Keeper’ residues and a nearby hydrophobic loop.
The Gate Keeper residues determine what nucleotides are allowed into the active site. In HIV-1 RT this is Tyrosine 115 (Y115) and in HERV-K RT it is Phenylalanine 126 (F126). The hydrophobic loop has a consensus sequence of 4 residues in most RTs. In HIV-1 RT this sequence is YMDD (Tyrosine-Methionine-Aspartic Acid-Aspartic Acid), while in HERV-K RT the Methionine is replaced by Isoleucine 195 (I195) giving the sequence YIDD. These two residue changes (Y115/F126 and YMDD/YIDD) have been mapped to HIV-1 RT drug resistance studies and have been shown to modulate potencies of NRTIs. In the case of the drug 3TC, which blocks HIV RT but is ineffective against HERV-K RT, its potency against HIV-1 RT is decreased 100-fold when the YMDD is changed to YIDD, which may be due to steric clashes that are introduced when the methionine is changed to isoleucine.
In addition, the change from YMDD to YIDD alters an allosteric inhibitor binding pocket that in HIV-1 RT can be blocked by non-nucleoside RT inhibitors (NNRTI’s). However, the I195 in HERV-K RT results in increased packing of this secondary site, reducing the space for NNRTIs to bind effectively, a likely explanation for why HIV NNRTIs do not block HERV-K RT.
Future Directions
Solving the X-ray structure of an HERV-K reverse transcriptase has provided insights into its activity and how to block it. This dimeric structure clarifies the reasons for poor inhibition of HERV-K RT by 3TC (lamivudine) and HIV NNRTI’s, despite significant similarities in the overall structure of HERV-K RT and HIV RT. Determining this structure is an important step in enabling the design of selective HERV-K RT tool compounds and inhibitors that could be brought forward in a number of disease areas.
Paul Wan, PhD, leads the Protein Sciences, Biophysics and Structural Biology Group at Charles River Early Discovery. He has over 25 years of experience working in the field of drug discovery in both an academic and commercial setting.
HERV-K: Solving the First Endogenous RT Protein Structure
Charles River Structural Biologists solved the first human endogenous retrovirus-k reverse transcriptase protein structure as part of a cross-function team led by ROME Therapeutics. Their findings appeared recently in PNAS.