
In the majority (90%) of ALS cases, disease onset is sporadic with the cause suggested to be a combination of genetic factors and exposure to environmental factors. In the remaining 10% of cases, ALS is caused by a specific genetic mutation. The genes most linked to ALS (both genetic and sporadic variants) are SOD1, TDP-43, FUS, and C9or72. These linked human genes enable the generation of transgenic animal models of ALS, and can be utilized in studies combining innovative fine kinematic gait analysis, motor behavior, preclinical imaging, and ex vivo biomarker and bioanalysis endpoints to investigate your ALS therapeutic in a disease-relevant model.
TDP-43 Mouse Model of ALS (rNLS8)
rNLS8 is an inducible, transgenic mouse model of ALS that is based on a human TDP-43 transgene. Overexpression of TDP-43 that lacks a nuclear localization signal is controlled by doxycycline administration. Removal of doxycycline from the diet results in expression and accumulation of hTDP-43 ΔNLS in the cytosol of motor neurons and induces disease onset. This TDP-43 animal model of ALS can be used to investigate the benefit of ALS therapeutics, including small molecules and advanced therapeutics, and has been validated using motor function tests, and tissue and fluid biomarkers.
Doxycycline is removed from the diet to induce an ALS phenotype in rNLS8 TDP-43 mice. Disease severity is monitored by body weight, hind limb clasping and tremor presence, with hind limb weakness apparent from seven days and significant changes in body weight two weeks after doxycycline removal. Rotarod and wire hang testing for motor coordination and strength, respectively, show dramatic reduction in motor function two weeks after stopping doxycycline treatment. Analysis of biomarkers of disease severity show increased neurofilament light chain (NfL) in CSF compared to controls, suggesting extensive axonal damage, as well as disintegration of the neuromuscular junction. Examination of cellular pathology shows increased total TDP-43 and phosphorylated TDP-43 in the spinal cord as well as cytosolic localization.
Phenotyping the rNLS8 ALS Model
Learn more about the phenotyping of the rNLS8 mouse model of ALS, including how our scientists measure disease severity in vivo and with ex vivo biomarker analysis.
View Poster
SOD1 Mouse Model of ALS (SOD1 G93A)
The SOD1 G93A mouse model of ALS expresses the G93A mutant form of human SOD1 and exhibits a similar phenotype to humans with limb-onset ALS. Disease severity and progression assessed by observation of climbing and walking shows onset of motor deficits usually from 13 to 15 weeks of age. Impairment in grip strength, wire hang, and rotarod performance are also seen at approximately the same age. Fine kinematic gait analysis enables investigation of minute changes in posture, gait, and limb movements and in the SOD1 mouse model is able to detect significant disease-related changes before the onset of visible changes in motor function, at 11 weeks of age or even earlier. Compound muscle action potential (CMAP) measurements show impaired motor neuron activity at the same time, also preceding observable muscle weakness. Reduced brainstem total volume can be identified in the SOD1 mouse model using MRI, and ex vivo analysis indicates motor neuron loss, degradation of the neuromuscular junction, and axonal damage (elevated NfL levels). Induction of neuroinflammatory responses can be identified through microglial activation (Iba1) and astrogliosis (GFAP) in the spinal cord.
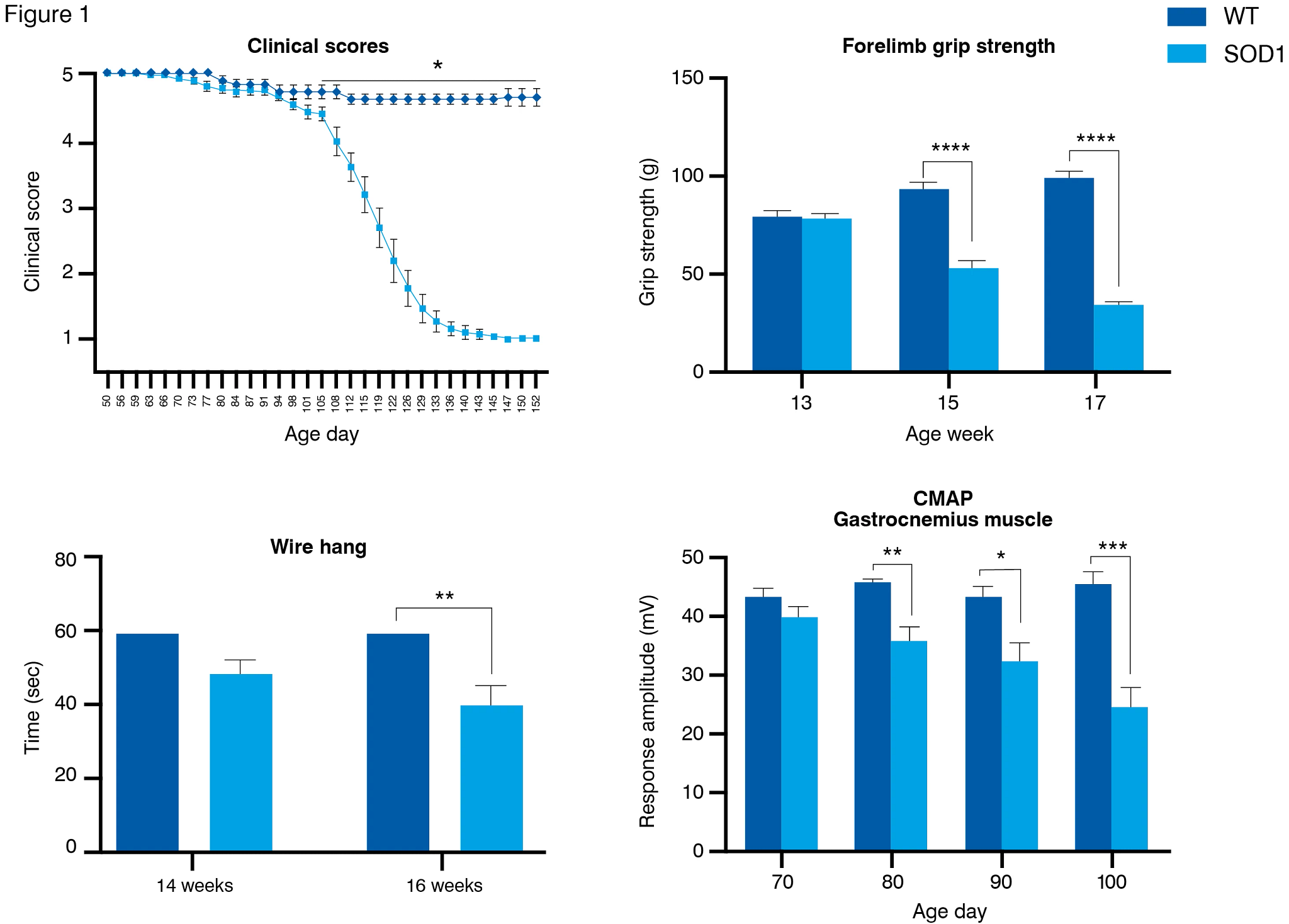
The SOD1 G93A mouse model of ALS displays onset of disease progression from around 13 weeks of age (A), with significant deficits in grip strength and wire hang tests from around 14 weeks of age (B and C). CMAP electrophysiology shows impaired motor neuron activity from 11 weeks of age.