
Featured News
Connecting with CADD: X-Ray Crystallography
Explore how X-ray crystallography and computational chemistry bind in this Q&A with Charles River experts.
Computational chemistry and structural biology have a long-standing synergistic relationship whereby structure determination of therapeutic targets, using techniques such as X-ray crystallography, followed by the application of computational chemistry methodologies can significantly accelerate the drug discovery process.
Following resolution of a protein-ligand structure, computer-aided drug design (CADD) can enable a deep-dive into the features of the binding pocket and create the opportunity to rapidly design and evaluate multiple alternative ligands against the binding site for compatibility. Scoring of the ligands, combined with the expertise of the computational chemist, is used to prioritize the most promising compounds for synthesis and experimental testing, hence CADD can expedite the hit identification, hit-to-lead, and lead optimization research phases.
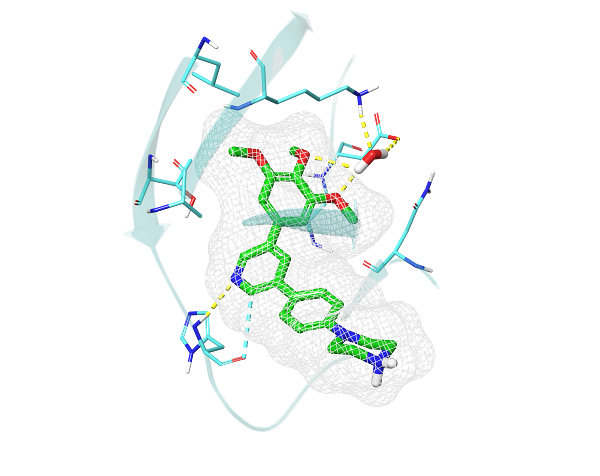
The crystal structure of Alk2 kinase in complex with LDN-213844 (4BGG). The cocrystalized inhibitor in green forms a hydrogen bond with the hinge as well as a water-mediated bond to the catalytic lysine.
We reached out to Paul Wan, Senior Group Leader of Protein Science, Structural Biology, and Biophysics, and Lampros Milanos, Senior Scientist in Computer-Aided Drug Design, to learn more about the relationship between structural biology and computational chemistry, and what can be achieved when these two sciences are brought together.
Why do you often recommend adding CADD into X-ray crystallography studies?
 Paul: The prime reason is that the CADD component corroborates high-quality crystallographic data. Using computer-aided drug design, we can provide additional quality controls focusing on the bound small molecule (if any) in relation to key residues in the protein. CADD plays a crucial role in complementing protein crystallography studies by providing computational insights into the interactions between proteins and small molecules, which can really accelerate drug discovery research.
Paul: The prime reason is that the CADD component corroborates high-quality crystallographic data. Using computer-aided drug design, we can provide additional quality controls focusing on the bound small molecule (if any) in relation to key residues in the protein. CADD plays a crucial role in complementing protein crystallography studies by providing computational insights into the interactions between proteins and small molecules, which can really accelerate drug discovery research.
What additional dimensions can CADD bring to structural biology research?
 Lampros: Computer-aided drug design approaches may differ from project to project, and hence diverse techniques can be applied in structural biology projects. The collaboration of the two disciplines can indeed confirm structure quality, and in addition can further advance projects through the use of various techniques aimed at water mapping, ligand binding mode stability assessment, fragment development, and binding site/mode analysis. These techniques can be essential for designing new analogues with improved binding and better properties.
Lampros: Computer-aided drug design approaches may differ from project to project, and hence diverse techniques can be applied in structural biology projects. The collaboration of the two disciplines can indeed confirm structure quality, and in addition can further advance projects through the use of various techniques aimed at water mapping, ligand binding mode stability assessment, fragment development, and binding site/mode analysis. These techniques can be essential for designing new analogues with improved binding and better properties.
How does CADD contribute to mapping water molecules and why is this important?
Lampros: Water molecule mapping can add in-depth understanding of the hydration properties of a protein binding site by computing position, entropy, and enthalpy of water molecules in the cavity. These values have many useful applications in drug design. They can, for example, be used to suggest modifications to a bound ligand which would displace or replace specific water molecules based on their free energy, often leading to improvements in binding affinity.
Paul: Water mapping helps identify water molecules that are critical for the stability and function of a protein's active site. These waters can mediate interactions between the protein and its ligands, often forming part of the binding interface. This data can provide insight into the thermodynamic contributions of water molecules to binding, and we can use this information to guide the design of drug candidates by suggesting modifications that can improve binding by optimizing protein interactions. Critically, water mapping provides a detailed understanding of the water environment within and around the target protein, enabling more informed and strategic decisions in the design and optimization of drug candidates.
What can CADD add to the understanding of ligand binding and its properties?
Paul: With protein ligand crystal structures, computer-aided drug design can help elucidate the structure-activity relationships (SAR) of compounds, providing insights into how structural modifications affect binding affinity and other pharmacological properties.
Lampros: Physicochemical properties and binding affinity of ligands are the essence of early drug discovery. The CADD team uses various methods to predict the binding free energy of the ligand (Free Energy Perturbation), test the stability of a ligand’s pose (Binding Pose MetaDynamics), or evaluate/rank interactions formed between the ligand and the protein (Fragment Molecular Orbital methods), and so on.
Regarding the physicochemical properties of ligands, Charles River has developed chemoinformatics tools and predictive models to generate predicted properties and ADME values, such as LogD, human intrinsic clearance, and tools to predict liabilities within the ligand structure.
Paul: This additional CADD knowledge can inform the design of new compounds and guide medicinal chemists and protein crystallography experiments to validate those structure-function relationships.
How can CADD support the development of a fragment hit?
Lampros: Our CADD software portfolio contains various tools that can support the development of fragment hits once the structure has been solved. A few techniques that can support fragment hits are library enumerations (reaction-based, R-group), hit expansion and ligand design, which introduce additional fragment groups into the fragment structure, developing it into a lead-like molecule with a molecular weight suitable for a small molecule drug.
What kind of structure-based projects are usually particularly amenable to adding a CADD step?
Paul: Most projects can benefit from CADD incorporation! Including projects that aren’t solely structure-based.
Lampros: That’s right. Accuracy of predictions may increase when a project is structure-enabled, as the protein’s binding site is also involved in the modeling, which provides information such as binding site shape and polarity/functionality that is just not available in a ligand-based approach.
On the other hand, as Paul mentioned, CADD can also contribute to ligand-based projects via several methods that have been successful in previous projects such as ab initio calculations, conformational searches, scaffold hopping, pharmacophore modeling, and molecular alignments. Furthermore, CADD can support antibody and target protein degradation projects (TPD). Once the complex of an antibody with an antigen has been experimentally determined, we can visualize and analyze the paratope-epitope interface and perform in silico modifications of selected amino acids aimed to improve the antibody stability or its affinity to the antigen. CADD can support TPD projects as well, by either modelling ternary complexes when structural information is available as well as degrader linker design using linker enumeration libraries.
How does the structural biology team work with researchers to design structure-based drug design workflows?
Paul: The term 'Structural Biology' encompasses a wide discipline, including protein sciences, biophysics, and what is seen as traditional structural biology (crystallography and Cryo-EM). Working with researchers, our teams rationally design optimal protein constructs for structural studies, and with these in place our biophysics teams can support screening of novel compounds coming out from a variety of hit finding strategies. The protein structures bound to the most promising compound candidate from biophysics triaging can be solved at the atomic level. The visualization of the interactions between the compound and the protein can help us in understanding and identifying binding sites and structural features crucial for drug interaction. Structural biology can guide the iterative process of lead optimization by providing feedback on the chemical modifications necessary to enhance the potency, specificity, and pharmacokinetic properties of lead compounds while minimizing off-target effects.
We take a very open and collaborative approach, which is really essential to ensuring we’re designing workflows that properly support the goals, downstream plans, and resources of our clients.
If you would like to learn more about how X-ray crystallography and / or computational chemistry can help build momentum into your drug discovery pipeline, please reach out to our team and talk to one of our drug discovery experts.