
Human iPSC-derived stem cell disease models from bit.bio (ioDisease Model Cells) are generated via opti-ox™ deterministic cell reprogramming technology and engineered to contain disease-relevant mutations, producing reliable and consistent disease models for a range of neurodegenerative and neuromuscular disorders. Once revived and cultured, these disease cell models can be used in phenotypic screening assays and high content imaging protocols, in lead optimization, and in vitro efficacy during drug development.
Huntington’s Disease Stem Cell Models
Huntington’s disease is a genetically inherited, autosomal dominant neurodegenerative disorder with symptoms characterized by motor, cognitive, and psychiatric deficits. The neuropathology is characterized by the dysfunction and death of striatal neurons, which is caused by a trinucleotide (CAG) repeat expansion in the Huntingtin (HTT) gene. bit.bio’s ioGlutamatergic Neurons HTT 50CAG/WT are deterministically programmed from human iPSCs and genetically engineered using CRISPR-Cas9 to carry a heterozygous 50CAG repeat expansion in exon 1 of the HTT gene.
Providing a robust and consistent iPSC-derived Huntington’s disease stem cell model, these cells show expression of pan neuronal and glutamatergic neuron markers from early on in differentiation. Multi-electrode array (MEA) analysis shows delayed neuronal network formation and decreased spontaneous activity in HTT 50CAG/WT neurons compared to genetically matched control cells. This stem cell disease model is suitable for use in phenotypic screening assays for the development of new therapies for Huntington’s disease, alongside genetically matched control cell lines.
Functional characterization of HTT 50CAG/WT neurons
Learn more about how HTT 50CAG/WT neurons have been validated by Charles River and how these disease cells can be used for screening of therapeutic compounds.
View poster
Amyotrophic Lateral Sclerosis (ALS) Stem Cell Disease Models
ALS is a neurodegenerative disease that weakens muscles and impacts physical function and is the most common form of motor neuron disease. Although most ALS patients have no family history, mutations in over 40 genes have been identified that contribute to the development of ALS, including in the TARDBP gene, which encodes transactivation DNA-binding Protein 43 (TDP-43). Mutations in TARDBP are also associated with frontotemporal dementia (FTD), and in both disorders cause increased protein phosphorylation, mislocalization and aggregation, and altered mRNA splicing of genes such as stathmin-2 (STMN-2).
ioGlutamatergic Neurons TDP-43 M337V/WT and ioGlutamatergic Neurons TDP-43 M337V/M337V are human iPSC-derived glutamatergic neurons genetically engineered to express heterozygous or homozygous M337V mutations in TDP-43. Providing a physiologically relevant ALS stem cell disease model, these can be used alongside genetically matched control glutamatergic neurons in phenotypic screening assays for development of new therapeutics for ALS.
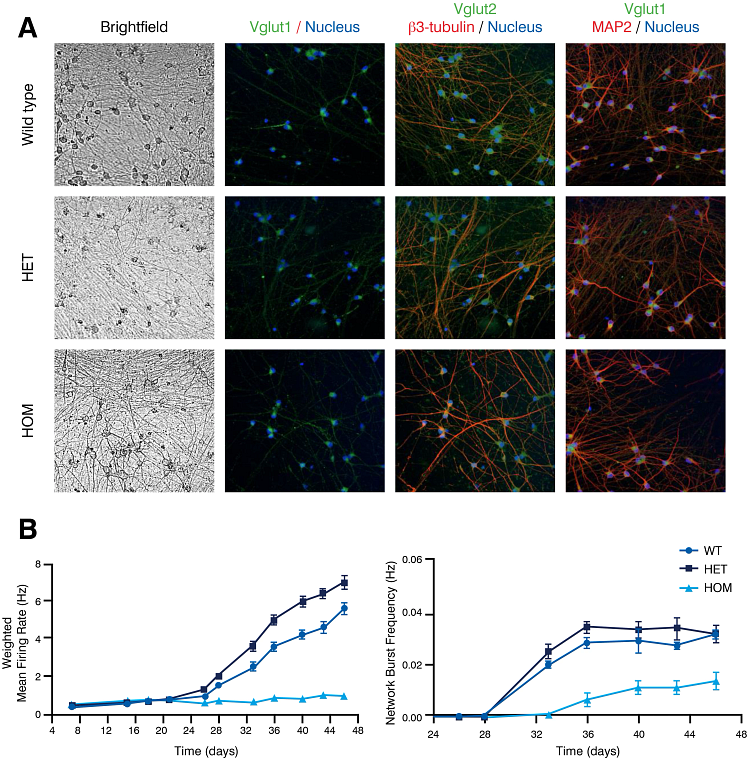
Figure 1: Expression of key neuronal markers and electrical activity analysis in ALS stem cell disease model. ioGlutamatergic Neurons TDP-43 M337V/WT (het) and ioGlutamatergic Neurons TDP-43 M337V/M337V (hom) show comparable expression of neuronal markers to their genetically matched wild-type controls (WT) (A). Neuronal activity, measured by mean firing rate and network burst frequency using multi-electrode array, was decreased in the TDP-43 M337V/M337V homozygous cell line, compared to the heterozygous and wild type cell lines.
Duchenne’s Muscular Dystrophy Stem Cell Disease Models
Duchenne’s muscular dystrophy (DMD) is an X-linked muscle weakness and degeneration disorder caused by mutations in the gene coding for dystrophin. Mutations resulting in single or multiple exon deletions are commonly identified in DMD patients with most mutations occurring in a hotspot region encompassing exon 45 to 55.
See how MEA electrophysiology can be used with primary cells, human iPSC-derived cells, and ex vivo brain slices across different stages of drug discovery in our on-demand webinar.
Watch Now
ioSkeletal Myoctyes carrying a hemizygous exon 44 or exon 52 deletion (ioSkeletal Myocytes DMD Exon 44 Deletion and ioSkeletal Myocytes DMD Exon 52 Deletion, respectively) are a disease-relevant human DMD stem cell model for drug discovery. These cells lack expression of dystrophin 10 days post-revival and can be paired with their isogenic wild-type cells as a control for use in phenotypic screening assays and high content imaging. Assessment of protein expression shows that dystrophin levels can be restored by treatment of DMD Exon 44 deletion myocytes with an exon 45 skipping antisense oligonucleotide (ASO).
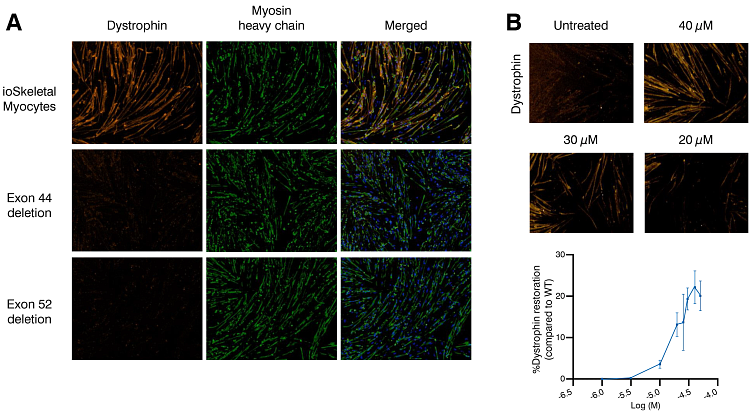
Figure 3: DMD stem cell disease models with exon 44 or exon 52 deletion lack expression of dystrophin when compared to genetically matched ioSkeletal myocytes (A). Treatment of ioSkeletal Myocytes DMD Exon 44 Deletion cells with an exon 45-skipping ASO resulted in a concentration-dependent restoration of dystrophin protein levels (B).
Custom Stem Cell Disease Models
Utilizing bit.bio’s opti-ox™ deterministic programming technology, custom disease stem cell models can be developed to fit your exact needs. Human iPSC-derived glutamatergic neurons, GABAergic neurons, microglia, and skeletal myocytes can be engineered to carry your mutation of choice, generating disease-relevant models to improve translatability in your drug development processes.

iPSCs for Research Use
With our iPSC portfolio, you have access to human-derived, wild-type iPSCs procured under Current Good Tissue Practice (CGTP) and conform to Global Ethical Standards and Clinical Cell Regulations. Learn more