In Vivo Models for Translation of Huntington’s Disease in Humans
We conduct contract in vivo Huntington’s disease animal studies to test the efficacy of novel therapeutics. We offer two mouse models, the zQ175 mouse model (JAX# 029928) and the transgenic R6/2 mouse model, as well as rat and large animal studies. All of our studies display robust, gene dose-dependent, progressive, and early-onset alterations in the validated endpoint assays. As part of our Neuroscience and Rare Disease portfolio, we also offer a selection of validated in vitro Huntington's disease assays as well.
-
Huntington's Disease Study Components
- Body weight
- Motor deficits
- Open field test
- Rear climbing test
- Rotarod test
- Grip strength test
- Cognitive deficits
- Procedural two-choice swim test
- Contextual fear conditioning
- Neurological index
- In vivo brain imaging
- MRI for brain volumetry (whole brain, cortex, striatum)
- MRS for striatal metabolites
- Biomarkers
- Various options in mRNA and protein detection assays
Exploratory Toxicology for Neuroscience Drug Discovery
This eBook describes strategies across the early stages of drug discovery to support confidence in your lead small molecule candidate and to ensure you proceed through the drug development process with the most promising candidate.
Download eBook
zQ175 Mouse Model
The zQ175 Mouse contains human mHTT allele with the expanded CAG repeat (~179 repeats) within the native mouse huntingtin gene. Thus, this animal model reflects an accurate representation of Huntington’s disease in humans from a genetic aspect. Both homo- and heterozygous zQ175 mice exhibit first signs of motor symptoms from 3-4 months of age. Compared to the R6/2 mouse model disease progression occurs slowly, making the zQ175 model more suitable for the assessment of chronic treatments.
Our neuroscience experts have established a phenotype of the zQ175 model, using motor tests and in vivo imaging methods, against which your novel therapeutics can be tested:
- Age-dependent changes in gait and subtle motor movements can be detected by detailed kinematic motor and gait analysis
- Tapered beam tests capture show specific motor deficits from 6 months of age
- MRI and MR spectroscopy show progressive brain atrophy and striatal metabolite changes, respectively, from 8 months of age
- PET imaging with dopamine receptor ligands shows decreased expression of D2/3 receptors in the striatum
- QuantiGene data shows changes in the expression of Huntington’s disease-related genes and HTT splice variants
-
zQ175 Mouse Model Example Data
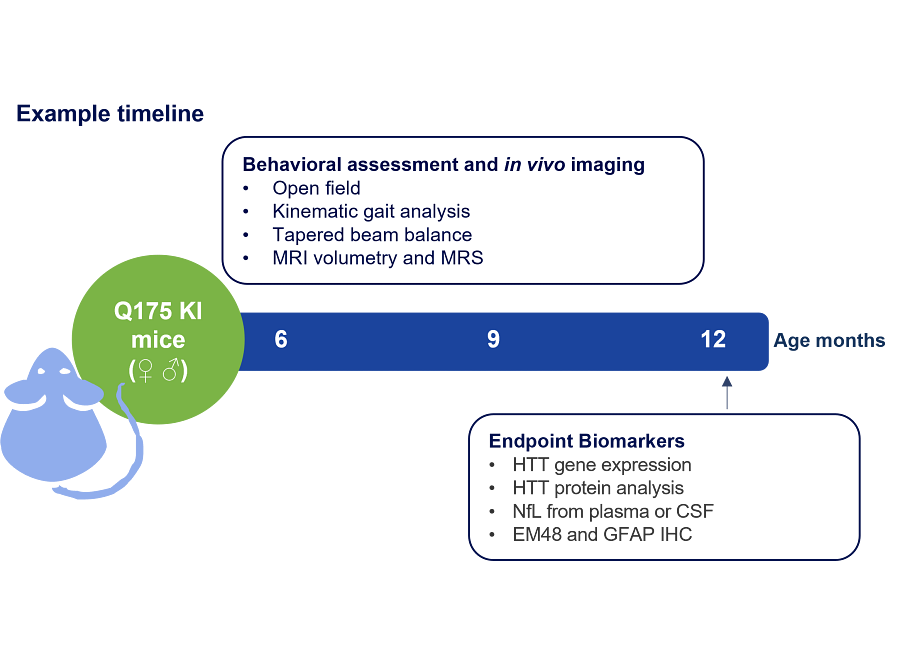
Figure 1: Example timeline of a zQ175 Huntington’s disease mouse study with recommended readouts and endpoint biomarkers.
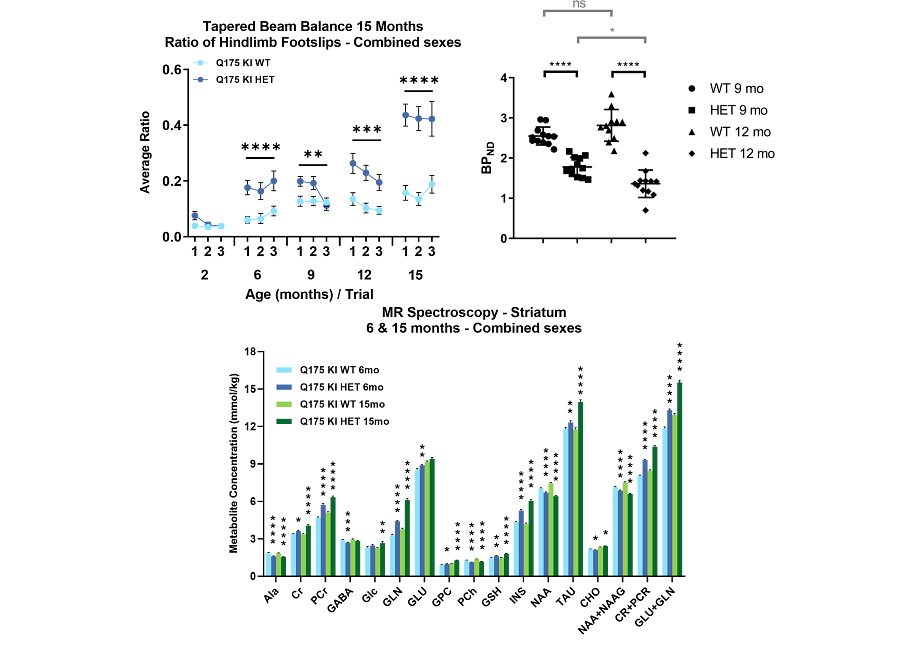
Figure 2: Example phenotypic profiling of zQ175 Huntington’s disease mice showing impairment from 6 months of age in the tapered beam balance, decreased expression of D2/3 dopamine receptors determined by PET imaging, and altered neurometabolite profile determined by MRS.
Transgenic R6/2 Mouse Model
Our most commonly used model is the transgenic R6/2 mouse model [B6CBA-Tg(HDexon1)62Gpb/1J], regularly used for proof of concept and efficacy studies. The R6/2 mouse contains N-terminally truncated mutant HTT (mHTT) with CAG repeat expansion (~125 repeats) within the huntingtin gene exon 1. This model rapidly develops a progressive phenotype of HD-like symptoms starting as early as 6-8 weeks of age:
- Decreased latency to fall on the Rotorod test
- Alterations in gait profile determined by kinematic gait profiling
- Decreased whole brain striatum and cortex volume measured by MRI
- Changes in neurometabolism determined by MR spectroscopy
- Expression of mutant HTT and neuroinflammatory markers
-
R6/2 Mouse Model Example Data
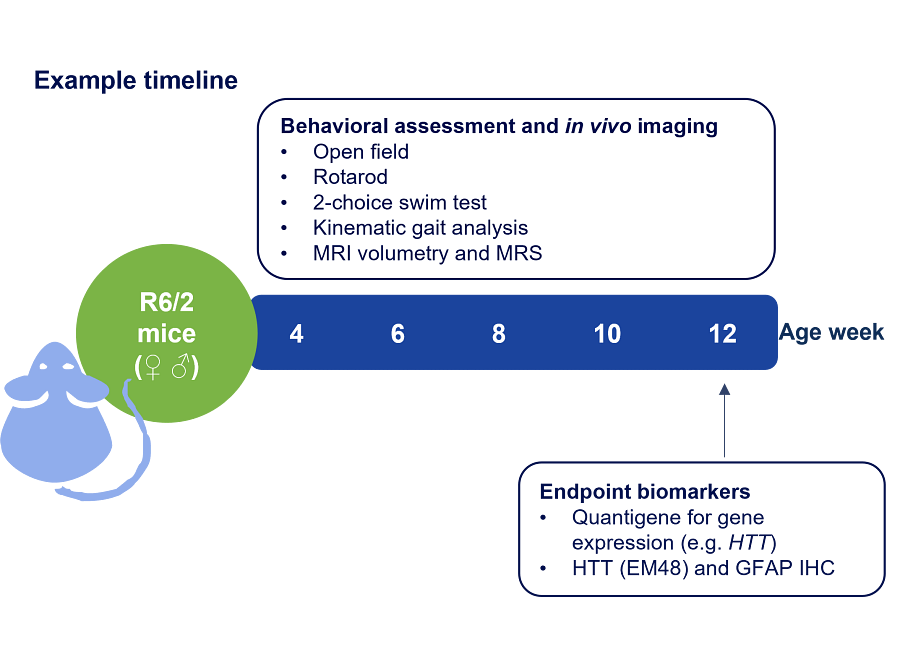
Figure 3: Example timeline of a R6/2 Huntington’s disease mouse study with recommended readouts and endpoint biomarkers.
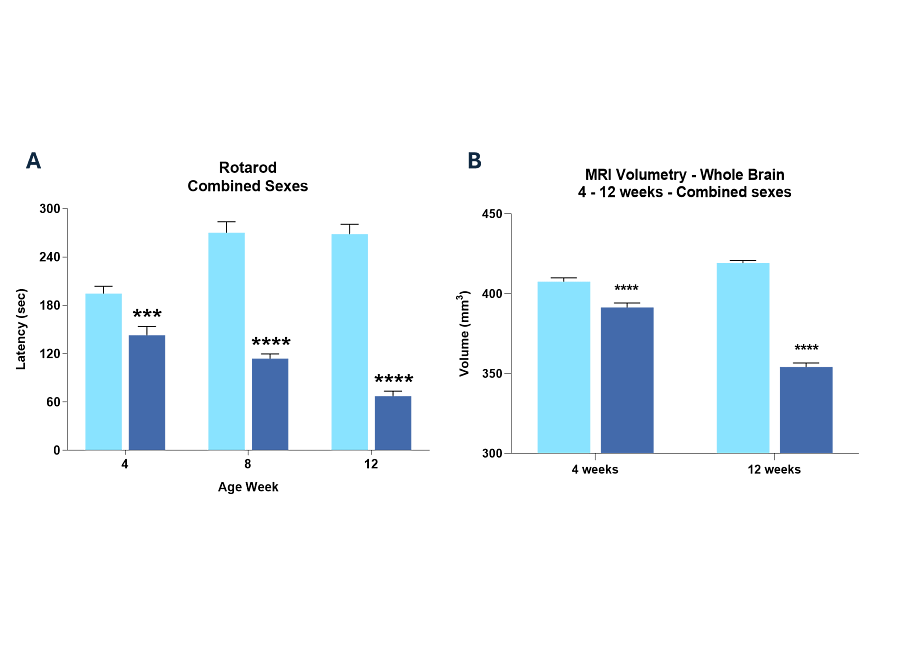
Figure 4: Example phenotypic profiling of R6/2 Huntington’s disease mice showing impairment in rotorod performance from 4 weeks of age, and changes in whole brain volume determined by MRI.
-
What factors are important for In Vivo Huntington’s disease animal studies?
When evaluating a potential therapeutic, it is important to have an animal model that accurately represents the neuropathology and symptomology of the disease. It is also crucial to have robust, gene dose-dependent, progressive and early-onset alterations in your study. We offer various customizable Huntington’s disease animal studies that take all of these factors into consideration.
-
What Huntington’s disease animal studies can I apply to my research?
Our models include the zQ175 mouse, the transgenic R/62 mouse, as well as rat and large animal models. We also offer validated in vitro studies.